- PRION BACKGROUND - Some information you may not yet
know about
-
- The prion diseases are a large group of related neurodegenerative
conditions, which affect both animals and humans. Included are Creutzfeldt-Jakob
disease (CJD) and Gerstmann-Sträussler-Scheinker (GSS) in humans;
bovine spongiform encephalopathy (BSE), or mad cow disease, in cattle;
chronic wasting disease (CWD) in mule deer and elk; and scrapie in sheep.
These diseases all have long incubation periods but are typically rapidly
progressive once clinical symptoms begin. All prion diseases are fatal,
with no effective form of treatment currently; however, increased understanding
of their pathogenesis has recently led to the promise of effective therapeutic
interventions in the near future. [1]
-
- There is also a variant CJD which has different pathological
effects as explained in [2].
-
- It is also noteworthy that Prion size was identified
back in 1971. Structures have both straight shapes and spirals. In the
photo below, a human strain is compared to hamster prion:
-
-
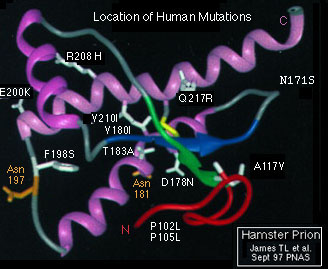
-
- Image credit:
- http://www.mad-cow.org/prion_structure_folder/human_mutsRIB_pnas.jpeg
-
-
-
- FREQUENCY OF INFECTIONS (number of cases per million
people):
-
-
- * In the US: The most common prion disease is CJD, with
a uniform incidence of approximately 1 case per million population both
in the United States and internationally. Familial forms of prion diseases,
such as GSS and fatal familial insomnia (FFI), are much more rare. About
10% of cases of CJD are familial, with an autosomal dominant pattern of
inheritance linked to mutations in the PRNP gene.
-
- * INTERNATIONALLY:
- So far, over 100 cases of NiCad have been reported in
the United Kingdom. Whether these patients represent the beginning of a
growing epidemic (such as that which occurred with BSE) or whether the
number of cases will remain relatively low remains unclear. For the past
2 years, incidence of nvCJD in the United Kingdom has remained stable,
with 27 cases reported in 2000 and 20 cases reported in 2001. Within the
first 6 months of 2002, 22 new cases have been recorded.
-
- Two populations are affected by CJD: Libyan-born Israelis
and some populations in restricted areas of Slovakia where the incidence
of CJD is 60-100 times greater than expected. These clusters were postulated
to be related to dietary exposure of the scrapie agent; however, this was
not supported by case-controlled studies. These local high rates of CJD
are linked to a high prevalence of codon 200 mutations in the PRNP gene.
The human PrP gene (PRNP) is found on chromosome 20.
-
- * The mean duration of sporadic CJD is 8 months.
-
- * nvCJD has a slightly longer course, with a mean duration
of 16 months.
-
- * Familial CJD has a mean duration of 26 months, while
GSS has the longest course, about 60 months.
-
- RACE:
-
- Sporadic CJD occurs throughout the world in people
of all races and typically has similar features.
-
- * Some familial forms of prion disease, such as familial
CJD, can have distinct features in an ethnic group. For example, familial
CJD in the Libyan Jewish population associated with a codon 200 mutation
has features of a peripheral neuropathy in addition to the more typical
manifestations of CJD (Meiner, 1992; Neufeld, 1992).
-
- * nvCJD has been limited to Europe, with almost all cases
occurring in the United Kingdom.
-
- SEX:
-
- No sex preponderance is known in prion diseases, with
some rare exceptions. For example, women had a greater tendency than men
to develop kuru because it was part of the ritual cannibalism for women
to eat the brains (and neural tissue has the highest dose of PrPSc).
-
- AGE:
-
- * The mean age of onset of sporadic CJD is 62 years.
The incidence of sporadic CJD is about 1 case per million population; however,
among individuals aged 60-74 years, the incidence is 5 cases per million
population (Holman, 1996). The age range can be broad; cases have been
reported in people as young as 17 years and as old as 83 years (Masters,
1979; Cathala, 1987).
-
- * nvCJD occurs in younger patients, with a mean age of
onset of 29 years.
-
- * Familial CJD, GSS, and FFI have mean ages of onset
ranging from 45-49 years. [1]
-
-
- WHY PRION PROTEINS ARE DIFFERENT FROM ORDINARY PROTEIN
- A Prion is a rod-like protein that grows more in length
than in diameter, penetrating and destroying brain tissue. Normal proteins
fold, while prions do not. The mechanism is not yet well understood in
the science community. Considerable research into prion has been taking
place at various institutions in the world. When Prisons infect humans,
it is called Creutzfeldt-Jakob Disease (pronounced "Crutz-felt Yah-cub",
or known as simply as CJD.) There is strong evidence today that at least
one strain of prions which infects cows, crosses the blood-brain barrier
in humans by eating infected meat.
-
- MORE VIRULENT THAN VIRUSES
- People have survived some of the deadliest, fastest
acting pathogens on earth such as Ebola which can kill in just 3 days.
Although not considered 100% fatal as the movies suggest survival is rare,
however. prions can withstand the high vacuum of space, standard sterilization
techniques and temperatures in excess of 1,000 degrees. CJD is considered
always to be 100% fatal. Recent news described the spread of the disease
in Florida via medical instruments. These instruments were used in operating
rooms on the surgery of 500 patients. Consider that this is just ONE known
case voluntarily made public. Statistics would dictate that this terrible
"accident" will already have happened across the world, in perhaps
or even thousands of times in many hospitals.
- The problem is most likely worse in third world countries,
where disposable medical instruments may be rare or non-existent.
-
- The Florida incident raises many troubling questions
such as:
-
- 1 .Why haven't thousands or millions of people died already
as a result of infected instruments and other causes ? Prion infections
are considered 100% fatal.
-
- 2. Could a Prion epidemic be successfully covered up
? There have a few families of CJD patients, who were told by the hospital
to keep quiet about the loss of a loved one. But when the number of CJD
victims reaches thousands, even this form of damage control will not work.
-
- 3. The existence of prions has been known about for many
years, and yet surgery continues with ineffective instrument sterilization.
And little is said about raising the Standard of Care that doctors and
hospitals follow to lower the risk of lawsuits. Why are hospitals not yet
using ALL disposable instruments that contact bodily fluids, especially
those instruments used for brain surgery ? One notices the safety glasses,
face shields and other protection doctors and nurses now use around patients.
Why are patients also not protected so carefully ?
-
- 4. In the UK it was found that prions are also present
in the tears of infected patients. Some optical instruments, such as a
physical contact-type Tonometer for measuring glaucoma, can transmit the
disease from one patient to another. The doctor or office staff must disinfect
the end of the Tonometer after each patient. But a survey showed this does
not happen with regularity. And disinfect it with WHAT ? Is there something
that is proven to work ? More advanced instruments used in the United States
use a puff of filtered air to measure eye pressure, without any physical
contact. It's amazing that physical contact Tonometers are still used.
At the very least, a sterile throw-away plastic cover on the device can
stop the spread of prions (CJD) and other eye diseases as well.
-
- 4. Do workers in meat and food processing factories
have a higher incidence of this disease than non-workers ? Does anyone
know ?
-
- TRANSMISSION PATHS TO HUMANS
- We must ask this question - have they always been here
and just now being recognized ? This seems very likely.
-
- All of us can be exposed to prions through various carriers
such as:
-
- * Medical and dental instruments - The current Standard
of Care does not require disposable medical instruments for ALL
- invasive medical procedures. You can be sure the penny-pinching
medical community will not do this until ordered to do so.
- This may or may not come in time.
-
- * Cat and dog food, commonly made from road kill and
sick animals, is only heated to about 250 degrees to process it.
-
- * Meat and other food made from cows that may be infected,
and were not detected (or looked for) in processing.
-
- * Lipstick, made from the brains of slaughtered cattle.
Since it is a cosmetic, this source of infection was never regulated until
- recently. It may also be in other cosmetics as yet unrevealed.
-
- * Contact with pets that eat infected pet food, but this
is vector not yet proven.
-
- There are other forms of transmission not covered above,
but these are enough.
-
- CRAZED WILDLIFE - RABIES OR MAD COW ?
-
- Ever see a mad deer or other animal struggle to stand,
or intentionally walk out on weak ice on a lake in early spring ? I've
witnessed this happen - after rescuers risk their lives to release the
animal from a certain deep freeze death, incredibly the animal immediately
runs back out on the same ice again. Clearly something is wrong. One may
not readily know whether the animal is in the last stages of rabies or
has prion disease, since both diseases attack the brain. Only by removing
the dead animal's head and sending it to a pathology lab can we know what
the affliction is. Many police departments have rabies kits, an unwanted
duty described to me by a local state police officer: The sick animal is
shot and the officer puts on rubber gloves. He then decapitates the animal
and places the head into a sealed plastic bag, along with the gloves. The
remains of the animal are buried, and the bag is labeled, packed in ice
and air shipped to a testing lab. He noted it is a most unpleasant duty
and he avoids it any cost. Not to mention the paperwork it requires. How
many mad cow type cases have been ignored over the years by less than publicly
minded law enforcement officers ? That number is probably higher than we
think.
-
- HAVE PRIONS ALWAYS BEEN HERE ?
- The current increase of these cases could be attributed
to perhaps a new awareness in the medical community, or, the disease was
not here before. We do know that prions were measured and detected as far
back as 33 years ago. No one seems to be exploring the idea that we have
been made susceptible to this horrific disease - one that has ALWAYS been
here. Legionaires disease is a good example of this - caused by a bacterium
present everywhere in ordinary earth. Yet until some 60 Legionaires died
at a hotel back in the seventies no one knew it existed, hence the name.
The same is true for cancer - how many millennia passed while humankind
has been on the earth, before uncontrolled cell growth was called cancer
?
-
- My theory (which I'm sure others may share) is that
prions have always been here on earth. In some unexplained way, humans
and animals may now be more susceptible to this pathogen than ever before.
This susceptibility is indicated by changes in chromosome 20 (from research
stated in the above extract.) Could it be there is something in our environment
that triggers susceptibility ? Perhaps certain microwave RF frequencies
such as those used by cell phones, may cause a protein mutation in those
individuals who are already susceptible. Few people realize that a cell
tower, with a typical power level of 100 WATTS per ANTENNA, with (four)
three antenna arrays for a total of 12 antennas, RADIATES ALMOST DOUBLE
THE POWER OF A TYPICAL 700 WATT MICROWAVE OVEN. Perhaps Prion susceptibility
may be a chemical effect, caused by a common food additive.
-
- Controlled scientific tests can determine what stimulates
Prion growth. Rodents and primates will probably need to be sacrificed
to accomplish this as a matter of performing the science needed to develop
some conclusions. This won't make animal rights activists happy to read
this. But these are the same people who, when they are ill and need an
operation to save their life, forget all about animals. They don't want
to die, and on that day no matter how many animals were sacrificed, it
will all be irrelevant to them. Animal research isn't cheap- an adult
Rhesus monkey today actually costs a researcher about $30,000.00. Why do
they cost so much ? Because after birth, it takes months of very carefully
feeding a monkey in an ultra-clean environment to raise it to the required
maturity. An adult monkey has the mentality of about a four year old child.
Funding such tests is another matter altogether, especially if the cause
of prion growth implicates a billion dollar industry such as telephony.
One can be assured that the telephony industry won't accept such results
silently.
-
- CJD type pathogens probably have always been here on
Earth, but are now being stimulated into a growth mode. Other incurable
diseases are quite useful for population control. For example, mycoplasmas,
not HIV are the cause AIDS. According to NIH Report #9, dated 1969 AIDS
it is not HIV as most think. HIV is an opportunistic infection and is the
disease doctors are told to look for. This is just another example of an
outdated Standard of Care. [3] By the time HIV is detected, a patient
is already mycoplasma infected. AIDS, prions and other pathogens assist
in very effective and almost unstoppable population control.
-
- The reader will notice the terms "CJD" and
nvCJD" used in this article. Please read the reference material given
below to better understand the difference in the two types of prion infections.
-
- Ted Twietmeyer is founder of data4science.net, which
looks at scientific phenomena from a different perspective. The website
also actively solicits volunteers to submit observations from around the
world about various phenomena at http://www.data4science.net
-
- REFERENCES
- [1] Science on prions at http://www.emedicine.com/neuro/topic662.htm
-
- [2] Variant CJD described http://www.bchealthguide.org/healthfiles/hfile55b.stm
-
- [3] Professor Don Scott and mycoplasmas - http://www.nexusmagazine.com/articles/mycoplasma.html
|